
scRNA-seq 的原始數(shù)據(jù)格式和目前大多數(shù) scRNA-seq 分析過程都基于 FASTQ 文件(或壓縮格式 fq.gz)。Illumina 平臺(tái)測序數(shù)據(jù)默認(rèn)生成 BCL 格式文件,可以通過 CellRanger mkfastq 進(jìn)行轉(zhuǎn)換。scRNAseq 的分析流程包括數(shù)據(jù)預(yù)處理、處理和擴(kuò)展下游分析(圖 4),其中數(shù)據(jù)預(yù)處理包括質(zhì)控、read 比對(duì)和表達(dá)量化;數(shù)據(jù)處理包括標(biāo)準(zhǔn)化、批次效應(yīng)校正、歸一化、特征選擇(HVG 選擇)、降維與聚類、細(xì)胞分型注釋、差異表達(dá)分析(DEGs)、可視化;擴(kuò)展下游分析包括擬時(shí)序、細(xì)胞間相互作用(CCI)、通路富集分析、基因調(diào)控網(wǎng)絡(luò)(GRN)等下游分析。整體來看,scRNAseq 分析方法層出不窮,沒有絕對(duì)完美適用于所有場景的方法,分析工具重要的是獲取生物學(xué)信息,難點(diǎn)在于選擇最合適的方法。本文中,我們將提出總結(jié)常見的單細(xì)胞轉(zhuǎn)錄組分析方法,并對(duì)其優(yōu)缺點(diǎn)和適用范圍提出建議。
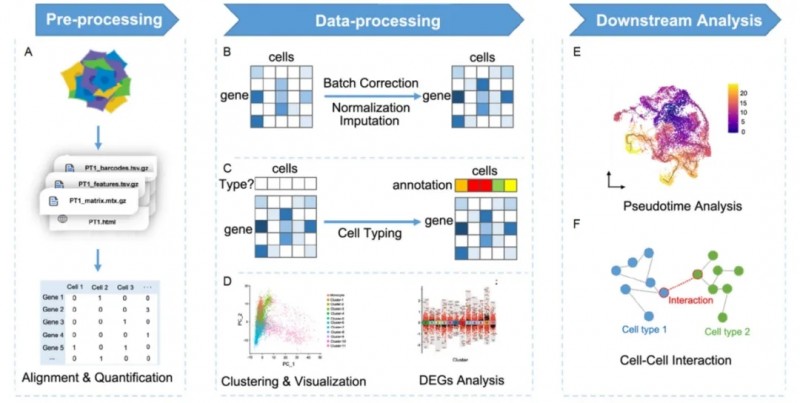
圖 4:單細(xì)胞分析概覽。A. 在預(yù)處理階段,基于測序數(shù)據(jù),細(xì)胞 - 基因矩陣讀數(shù)通過單細(xì)胞讀數(shù)校正和定量產(chǎn)生;B. 分析使用的高質(zhì)量細(xì)胞矩陣通過原始的基因表達(dá)矩陣獲得,通過去批次效應(yīng)矯正批次,通過標(biāo)準(zhǔn)化降低生物學(xué)差異,補(bǔ)充未檢測到的基因;C. 依照或不依照先前的參考信息對(duì)細(xì)胞類型進(jìn)行注釋;D. 轉(zhuǎn)錄組特征相似的細(xì)胞被歸為一類,稱為“細(xì)胞簇(cluster)”,細(xì)胞的可視化通過降維方法實(shí)現(xiàn),差異基因分析對(duì)組間差異進(jìn)行檢驗(yàn);E. 擬時(shí)序分析重建細(xì)胞轉(zhuǎn)錄水平變化的動(dòng)力學(xué)過程;F. 細(xì)胞間轉(zhuǎn)錄組調(diào)控關(guān)系可以通過胞間互作分析進(jìn)行推斷。
數(shù)據(jù)預(yù)處理
將原始測序數(shù)據(jù)通過濾除低質(zhì)量 reads 和環(huán)境干擾與參考基因組進(jìn)行比對(duì)和量化。從而得到每個(gè)細(xì)胞的特征計(jì)數(shù)矩陣和記錄其他信息的輔助文件,用于下游的數(shù)據(jù)分析(圖 4 A)。
1、質(zhì)控
由于測序儀器問題、人為操作、細(xì)胞自發(fā)情況,或存在空液滴、雙細(xì)胞、死細(xì)胞等,不可避免地會(huì)產(chǎn)生低質(zhì)量的測序數(shù)據(jù)(Chen 等,2019a ; Hao 等,2021b)。空液滴通常出現(xiàn)在液滴捕獲細(xì)胞外背景轉(zhuǎn)錄本而不是細(xì)胞時(shí)(Ilicic 等,2016 ; Kolodziejczyk 等,2015)。一種高度主觀的方法是根據(jù)曲線的膝點(diǎn)確定一個(gè) UMI 閾值,并過濾掉 UMI 計(jì)數(shù)低的細(xì)胞。隨后使用 DropEst (Petukhov 等,2018)、EmptyDrops (Lun 等,2019) 和 DIEM (Alvarez 等,2020) 增強(qiáng)過濾效果。DropletQC (Muskovic and Powell, 2021) 量化未剪接前 mRNA 含量的核分?jǐn)?shù)得分。MT 基因閾值雖然是衡量死細(xì)胞的標(biāo)準(zhǔn),但它的選擇需要綜合考慮細(xì)胞生理因素 (Subramanian 等,2022)。近年來,基于深度學(xué)習(xí)的方法也應(yīng)運(yùn)而生,例如基于神經(jīng)網(wǎng)絡(luò)的 EmptyNN (Yan 等,2021) 和基于深度生成模型的 CellBender (Fleming 等,2019),能夠有效識(shí)別空液滴中的背景轉(zhuǎn)錄本。
雙細(xì)胞是指兩個(gè)細(xì)胞包含在一個(gè)液滴中的情況,根據(jù)轉(zhuǎn)錄分布可分為同源雙峰和異源雙峰,均服從泊松統(tǒng)計(jì)量(Bloom, 2018)。絕大多數(shù)方法基于基因表達(dá)計(jì)算,利用先驗(yàn)知識(shí)或深度學(xué)習(xí)獲取單峰與雙峰細(xì)胞的差異,然后訓(xùn)練分類器進(jìn)行篩選,例如基于最近鄰的 DoubletFinder (McGinnis 等,2019a)、Scrublet (Wolock 等,2019);基于反卷積的 DoubletDecon (DePasquale 等,2019)、基于變分自編碼器的 Solo (Bernstein 等,2020) 和基于集成算法的 Chord (Xiong 等,2021a)。此外,Scds 是另一種篩選方法,它依賴于基于共表達(dá)的雙聯(lián)體打分和基于二分類的雙聯(lián)體打分策略,實(shí)現(xiàn) scRNA-seq 表達(dá)數(shù)據(jù)的雙聯(lián)體分離 (Bais and Kostka,2020)。一些方法使用其他特征,例如 demuxlet 它使用自然遺傳變異信息指導(dǎo)實(shí)驗(yàn)并通過計(jì)算進(jìn)行過濾 (Kang 等,2018)。
合理的質(zhì)控需要綜合考慮技術(shù)性和生物性因素,這也是當(dāng)前研究的主要方向。最近一種由生物數(shù)據(jù)驅(qū)動(dòng)的自學(xué)習(xí)無監(jiān)督質(zhì)控方法 ddqc 被提出來,用于確定各種 GC 指標(biāo)的具體閾值 (Macnair and Robinson,2023)。
2、reads 比對(duì)和定量
質(zhì)控后剩余的高質(zhì)量細(xì)胞需要將這些短 reads 映射到特定的參考基因組上進(jìn)行比對(duì),以此對(duì)基因表達(dá)水平進(jìn)行定量。RNA 比對(duì)通常分為兩步:比對(duì) reads 以建立索引和映射 RNA 剪接序列,前一步與 DNA reads 比對(duì)共用,解決錯(cuò)配問題并設(shè)置索引參考;后一步是 RNA reads 比對(duì)所特有的,提供連通性信息。
早期二代測序結(jié)果是幾十對(duì)長度的堿基 reads。Seed-to-extend (Buhler,2001)(包括 MAQ (Li 等,2008a)、SOAP (Li 等,2008b)、CloudBurst (Schatz,2009)、ZOOM (Lin 等,2008))、BurrowsWheeler 變換方法 (Burrows and Wheeler,1994)(包括 SOAP2 (Li 等,2009)、Bowtie (Langmead 等,2009)、BWA (Li and Durbin,2009))、Needleman-Wunsch 方法(包括 Novocraft (Hercus,2009))和 suffix-tree 算法方法(包括 MUMmer 2 (Delcher 等,2002))都是百萬級(jí)短鏈 DNA 測序 reads 比對(duì)的有效工具。Bowtie 采用了一種依賴于 Burrows-Wheeler Transforming 的 FM-index 方法,如果 reads 有多個(gè)準(zhǔn)確匹配則結(jié)果只報(bào)告一個(gè),與 MAQ(Ferragina and Manzini,2001)相比,大大優(yōu)化了運(yùn)行內(nèi)存和比對(duì)速度。BWA 是另一種基于 BWT 的比對(duì)方法,使用新的 SAM(Sequence Alignment/Map)格式輸出比對(duì)結(jié)果。基于 MAQ 和 Bowtie 兩種短鏈 DNA 比對(duì)算法,Cole Trapnell 于 2009 年提出了第一個(gè)針對(duì) NGS 數(shù)據(jù)的 RNA-seq 比對(duì)方法 TopHat,它使用 2 -bit-per-base 編碼實(shí)現(xiàn) reads 與哺乳動(dòng)物基因組中剪接位點(diǎn)的有效比對(duì),而無需事先知道剪接位點(diǎn)的具體信息(Trapnell 等,2009)。
上述方法在堿基對(duì)長度超過 50 bp 時(shí)比對(duì)精度急劇下降(Gupta 等,2018 ; Lebrigand 等,2020)。NGS 單細(xì)胞測序分析主要采用兩類方法:基于 Bowtie2 的方法和基于 seed 策略的方法(Langmead and Salzberg, 2012)。Bowtie2 是 Bowtie 的升級(jí)版,保留了 FM-index 依賴的 BWT 算法核心,允許有間隙比對(duì),并使用單指令多數(shù)據(jù)(SIMD)擴(kuò)展到長測序比對(duì),同時(shí)提高運(yùn)行速度。Daehwan Kim 在 Bowtie2 基礎(chǔ)上,先后提出了 TopHat2(Kim 等,2013)和 HISAT(Kim 等,2015)。種子策略主要有 STAR(Dobin 等,2013)和 Subread(Liao 等,2013)。STAR 基于最大可映射前綴(MMP)的思想,采用順序檢索的策略,將與參考匹配的最長部分 reads 設(shè)為種子 1,其余 read 繼續(xù)匹配,依次從種子 2 調(diào)用至種子 n。值得注意的是,Rsubread 完全基于 R 語言平臺(tái)實(shí)現(xiàn)了第一次 read 比對(duì)和基因量化的過程(Liao 等,2019)。
基因表達(dá)量化又可分為偽比對(duì)量化和基于 read 比對(duì)的量化。偽比對(duì)是指不采用上述嚴(yán)格的兩步法將所有 reads 比對(duì)到參考基因組上,包括選定的 k-mers 比對(duì)方法(Sailfish(Patro 等,2014)、Kallisto(Bray 等,2016)、Salmon(Patro 等,2017)、RapMap(Srivastava 等,2016)和 Barcode-UMI-Set (BUS) 比對(duì)方法 BUStools(Melsted 等,2019)。Kallisto-BUStools 是最新的工作流程,它使用 BUS 文件格式進(jìn)行初始數(shù)據(jù)預(yù)處理,與 BUStools 一樣,偽比對(duì)結(jié)果和量化計(jì)數(shù)都保存在 BUS 文件中(Melsted 等,2021)。另一方面,基于 reads 比對(duì)的方法依賴于 RNA reads 比對(duì)方法的結(jié)果來量化基因。CellRanger 是 10x Genomic 公司指定替代 Longranger 的官方開源數(shù)據(jù)預(yù)處理軟件(Zheng 等,2017)。STARsolo 是替代 Cellranger 的 mapping/quantification 功能的工具,可實(shí)現(xiàn)多平臺(tái)測序數(shù)據(jù)的分析和基因表達(dá)之外的轉(zhuǎn)錄組特征的量化(Kaminow 等,2021)。其他基于 reads 比對(duì)的基因表達(dá)定量方法如 UMItools (Smith 等,2017)、zUMIs (Parekh 等,2018)、Alevin-fry (He 等,2022)、DropEst (Petukhov 等,2018)、RainDrop (Niebler 等,2020)、baredSC (Lopez-Delisle and Delisle, 2022)、BCseq (Chen and Zheng, 2018) 使用各種質(zhì)量過濾器和 barcode/UMI 處理策略在一定程度上提高了 CellRanger 的性能。
CellRanger 和 STARsolo 在處理包括 10x Chromium 在內(nèi)的各種單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)集時(shí)都具有良好的運(yùn)行速度,并且準(zhǔn)確度極高。但在獲得幾乎相同結(jié)果的前提下,后者相比前者提升了至少 5 倍的運(yùn)行速度,這也驗(yàn)證了 Alexander Dobin 等人使用 STARsolo 取代 CellRanger 的目的(Brüning 等,2022 ; Chen 等,2021a ; You 等,2021)。
數(shù)據(jù)處理
在對(duì)表達(dá)矩陣進(jìn)行必要的調(diào)整(Normalization、Batch Effect Correction、Imputation)后,即可從單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)中充分挖掘出生物信息進(jìn)行分析。Seurat 和 Scanpy 分別基于 R 和 Python 對(duì)上述過程進(jìn)行模塊化、可擴(kuò)展的處理,是目前單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)的主流分析流程(Satija 等,2015 ; Wolf 等,2018)。常規(guī)分析流程和預(yù)期處理結(jié)果可參見總分析框架(圖 4 B-D)。
1、標(biāo)準(zhǔn)化
在測序過程中,技術(shù)原因或者細(xì)胞本身的生物學(xué)差異可能造成同一樣本內(nèi)(細(xì)胞之間)或者不同樣本之間的文庫大小差異(Marinov 等,2014)。無限數(shù)方法按照文庫大小進(jìn)行處理,按照具體原理大致可以分為基于全局縮放的標(biāo)準(zhǔn)化、spike-in 標(biāo)準(zhǔn)化和其他數(shù)據(jù)變換模型標(biāo)準(zhǔn)化。
全局縮放方法最初是為 bulk RNA 分析而發(fā)展起來的,通過特定的縮放因子對(duì)全局?jǐn)?shù)據(jù)進(jìn)行縮放(Finak 等,2015)。每萬計(jì)數(shù)(CPT)變換和每百萬計(jì)數(shù)(CPM)變換是常見的線性縮放方法,在不考慮 spike-in count 的情況下,它們都對(duì)每個(gè) UMI/ 總 UMI count 等距縮放。其他標(biāo)準(zhǔn)化方法包括每百萬 reads 數(shù)(RPM)(Mortazavi 等,2008)、修剪均值 M 值(TMM)、DESeq(Robinson and Oshlack, 2010)、上四分位縮放(Bullard 等,2010)、FPKM(Trapnell 等,2010)、RPKM(Tu 等,2012)等,它們對(duì)于極值的穩(wěn)定性比線性縮放更好,因此與 RPKM/FPKM 一樣具有更廣泛的應(yīng)用范圍。但單獨(dú)使用該類方法進(jìn)行單細(xì)胞轉(zhuǎn)錄組的標(biāo)準(zhǔn)化時(shí),由于數(shù)據(jù)的稀疏性和假陽性率虛高,效果并不可接受(Evans 等,2018),與特定方法結(jié)合時(shí)往往需要改進(jìn)。SCnorm 使用分位數(shù)回歸方法來評(píng)估不同測序深度依賴細(xì)胞組之間的尺度因子(Bacher 等,2017)。bayNorm 基于基因原始計(jì)數(shù)與真實(shí)計(jì)數(shù)服從負(fù)二項(xiàng)(NB)分布的假設(shè),使用集成貝葉斯模型對(duì) scRNA-seq 數(shù)據(jù)進(jìn)行標(biāo)準(zhǔn)化(Tang 等,2020)。
spike-in 標(biāo)準(zhǔn)化方法可以看作是全局尺度方法的另一種擴(kuò)展,因?yàn)槌叨纫蜃邮歉鶕?jù) spike-in 基因計(jì)算出來的。需要注意的是,將 RNA spike-ins 的信息添加到其他方法中也可以提高 SCnorm 等標(biāo)準(zhǔn)化的效果。GRM 是一種基于 spike-in ERCC 分子濃度伽馬分布的方法,其中 ERCC 是測序中常用的校準(zhǔn)材料(Ding 等,2015)。BASiCS 是一種自動(dòng)貝葉斯標(biāo)準(zhǔn)化方法,將泊松分層模型應(yīng)用于 spike-in(技術(shù))基因,以推斷細(xì)胞特定的標(biāo)準(zhǔn)化常數(shù)(Vallejos 等,2015)。
以上方法都是在細(xì)胞內(nèi) RNA 數(shù)量恒定的假設(shè)下對(duì)基因進(jìn)行縮放,而這可能具有欺騙性,因此其他轉(zhuǎn)化模型采用了不同的策略。由于單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)存在零膨脹問題,一些模型就是為此而設(shè)計(jì)的,例如相對(duì)對(duì)數(shù)表達(dá)(RLE)方法 ascend(Senabouth 等,2019)和基于 NB 的模型,如 Dino(Brown 等,2021)、scTransform(Hafemeister and Satija, 2019)。其他轉(zhuǎn)化模型歸一化方法如 MUREN 使用最小二乘(LTS)回歸算法(Feng and Li, 2021);Sanity 使用從 UMI 計(jì)數(shù)推斷出的對(duì)數(shù)轉(zhuǎn)錄商(LTQ)作為貝葉斯框架的輸入,以避免泊松波動(dòng),因?yàn)?LTQ 向量的變化估計(jì)了基因表達(dá)值(Breda 等,2021);PsiNorm 是一種基于無監(jiān)督帕累托分布尺度參數(shù)的方法,用于提升標(biāo)準(zhǔn)化效率和準(zhǔn)確率(Borella 等,2021)。Charles Wang 比較了 sctransform、TMM、DESeq 等共 8 種標(biāo)準(zhǔn)化方法,其中 sctransform 和 logCPM(Seurat 的內(nèi)置處理方法)受數(shù)據(jù)影響最小,在可變數(shù)據(jù)集上最穩(wěn)定(Chen 等,2021a)。
2、批次效應(yīng)校正
由于實(shí)驗(yàn)設(shè)計(jì)、測序平臺(tái)、測序時(shí)間、人員操作流程等原因,不同的單細(xì)胞轉(zhuǎn)錄組測序數(shù)據(jù)在 mRNA 捕獲效率、測序深度等會(huì)存在明顯差異,從而產(chǎn)生樣本間的批次效應(yīng)(Chen 等,2019a;Hwang 等,2018;Tung 等,2017)。理論上可以通過實(shí)驗(yàn)策略消除技術(shù)變異,但由于實(shí)驗(yàn)過程的客觀限制以及測序儀器誤差,不可避免地會(huì)或多或少地引入批次效應(yīng)。利用計(jì)算方法進(jìn)行校正是解決不完善實(shí)驗(yàn)設(shè)計(jì)的必要手段,通常使用的方法可以分為相互最近鄰(MNN)方法、基于潛在空間的方法、基于圖的方法、DL 方法和其他方法。
MNN 首先識(shí)別出不同批次之間同一細(xì)胞類型的最相似細(xì)胞,然后利用這些細(xì)胞進(jìn)行批次效應(yīng)校正,包括 batchelor(Haghverdi 等,2018)、Scanorama(Hie 等,2019)、Canek(Loza 等,2022)。另一類使用 MNN 的方法是基于降維后的潛在空間,如 Seurat (Satija 等,2015)、BEER (Zhang 等,2019b)、SMNN (Yang 等,2021a)、iSMNN (Yang 等,2021b)。例如,Seurat 使用典型相關(guān)分析 (CCA) 潛在空間中的 MNN 對(duì) (稱為“錨點(diǎn)”)來匹配相似細(xì)胞,而 BEER 使用主成分分析 (PCA) 子空間來篩選相似性較差的子群。SMNN 和 iSMNN 分別采用監(jiān)督機(jī)器學(xué)習(xí)和迭代監(jiān)督機(jī)器學(xué)習(xí)來細(xì)化從預(yù)校正細(xì)胞聚類或迭代細(xì)胞聚類信息中訓(xùn)練出的 MN 對(duì)。
基于潛在空間的方法是指在隱藏空間或降維后的嵌入中進(jìn)行批次效應(yīng)校正的方法,除了基于 MNN 聚類的策略外,還有與 PCA 相關(guān)的空間方法 Harmony(Korsunsky 等,2019)、FIRM(Ming 等,2022)、Monet(Wagner, 2020);t 分布隨機(jī)鄰域嵌入 (tSNE) 空間方法 sc_tSNE(Aliverti 等,2020)和 ZINBWaVE(Gao 等,2019)。Harmony 被廣泛用于去除樣本間的批次效應(yīng),使用 PCA 方法將排序的細(xì)胞輸入到單個(gè)公共嵌入中,然后在最大多樣性聚類和線性批次校正之間迭代循環(huán),直到為每個(gè)細(xì)胞分配一個(gè)特定的校正因子,可用于后續(xù)的批次效應(yīng)去除。Sc_tSNE 方法引入梯度下降算法對(duì)傳統(tǒng) t -SNE 算法進(jìn)行優(yōu)化,隨后采用線性校正(Aliverti 等,2021)。ZINB-WaVE 最初設(shè)計(jì)用于在單細(xì)胞數(shù)據(jù)中進(jìn)行基因提取,Risso et al.(2018)將該方法擴(kuò)展至小批量優(yōu)化。
基于圖的方法利用細(xì)胞基因表達(dá)矩陣將數(shù)字信息轉(zhuǎn)化為空間構(gòu)造的圖,其中節(jié)點(diǎn)代表不同類型的批次,邊的權(quán)重基于不同的計(jì)算方法。BBKNN 利用 k 近鄰細(xì)胞構(gòu)建圖(KNN 圖),通過使用均勻流形近似與投影(UMAP)方法合并不同數(shù)據(jù)集間單個(gè)細(xì)胞的圖實(shí)現(xiàn)批次效應(yīng)校正,這也是 Scanpy 工作流程中的默認(rèn)方法(Pola ński 等,2020 ; Wolf 等,2018)。王波在 OCAT 中提出“幽靈細(xì)胞”(默認(rèn)為 k-means 算法聚類中心)來制作細(xì)胞連接的二分圖(Wang 等,2022a)。
近年來,深度學(xué)習(xí)方法的快速發(fā)展也為批次效應(yīng)校正提供了新思路,實(shí)現(xiàn)高效、大通量的數(shù)據(jù)處理,如 INSCT(Simon 等,2021)(三重態(tài)神經(jīng)網(wǎng)絡(luò))、CLEAR(Han 等,2022)(自監(jiān)督對(duì)比學(xué)習(xí))、BERMUDA(Wang 等,2019e)(遷移學(xué)習(xí))、iMAP(Wang 等,2021a)(VAE-GAN)、ResPAN(Wang 等,2022e)(Wasserstein GAN),一些新方法被證明在批次效應(yīng)校正方面有更好的效果;例如,基于從 SciBet 學(xué)習(xí)到的帶注釋數(shù)據(jù)集中的生物學(xué)先驗(yàn)知識(shí),SSBER 可以在大型 RNA 測序數(shù)據(jù)集中去除批次效應(yīng)(Zhang and Wang,2021)。建議在整合單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)之前,應(yīng)根據(jù)數(shù)據(jù)的實(shí)際情況先測試多種方法,然后選擇最合適的批次效應(yīng)去除方法。例如,Jinmiao Chen 團(tuán)隊(duì)和 Charles Wang 團(tuán)隊(duì)分別于 2020 年和 2021 年對(duì)本綜述 2.2 中提到的前三種方法的大部分進(jìn)行了基準(zhǔn)測試,證明了 Harmony 和 Seurat V3 在大多數(shù)情況下都能達(dá)到良好的批次效應(yīng)校正效果,這符合這兩種方法如今仍然被廣泛使用,但對(duì)于深度學(xué)習(xí)方法來說仍然缺乏好的指標(biāo)這一事實(shí)(Chen 等,2021a;Tran 等,2020)。
3、填補(bǔ)
測序過程中會(huì)引入大量 0 值(高通量大規(guī)模 10x 基因組測序數(shù)據(jù)中零值可能超過 90%)(Stegle 等,2015 ; Talwar 等,2018),這會(huì)干擾下游生物學(xué)差異分析,因此必須對(duì)原始基因表達(dá)矩陣中的缺失數(shù)據(jù)值進(jìn)行填補(bǔ),同時(shí)有效區(qū)分技術(shù)噪音零值與生物學(xué)零值。
基因 / 細(xì)胞分離方法主要應(yīng)用于早期的填補(bǔ),其分別考慮細(xì)胞相似性(MAGIC (van Dijk 等,2018)、Sclmpute (Li and Li, 2018)、VIPER (Chen and Zhou, 2018)、RESCUE (Tracy 等,2019)、scRMD (Chen 等,2020a)、scRoc (Ran 等,2020))或基因間關(guān)系(SAVER (Huang 等,2018a)、SAVER-X (Wang 等,2019a)、G253 (Wu 等,2021e)、DCA (Eraslan 等,2019)、DeepImpute (Arisdakessian 等,2019))。總體而言,這些方法缺乏對(duì)數(shù)據(jù)集整體的考慮,容易導(dǎo)致過度插補(bǔ)或者引入誤差(Zhang 等,2019d)。綜合方法綜合考慮細(xì)胞與基因之間的聯(lián)系:CMF-Impute 和 netNMF-sc 是最早有效利用細(xì)胞與基因之間的關(guān)聯(lián)進(jìn)行插補(bǔ)的方法(Elyanow 等,2020;Xu 等,2020a)。scIGANs 通過特定的 GAN 模型處理基因表達(dá)矩陣,利用生成的細(xì)胞訓(xùn)練 GANs 模型來插補(bǔ) dropout(Xu 等,2020b)。近年來,新的方法還在不斷被提出,以更好地解決 dropout 之外的技術(shù)噪聲對(duì)數(shù)據(jù)的影響,并實(shí)現(xiàn)對(duì)生物零值的更好的區(qū)分。AutoClass(Li 等,2022c)實(shí)現(xiàn)了無監(jiān)督處理,而 ALRA 方法主要針對(duì)生物零值(Linderman 等,2022)。scMOO 進(jìn)行了根本性的改變,利用數(shù)據(jù)的潛在結(jié)構(gòu)來學(xué)習(xí)細(xì)胞相似性垂直結(jié)構(gòu)和總低秩結(jié)構(gòu)中的深度關(guān)聯(lián),從而取得了比單一基因表達(dá)矩陣作為輸入更好的插值效果,但對(duì)內(nèi)存的要求也更高(Jin 等,2022a)。sc-PHENIX 利用 PCA-UMAP 初始化方法,實(shí)現(xiàn)了基因表達(dá)的非線性插值(Padron-Manrique 等,2022),目前哪種插值能取得最佳效果尚無明確定論。由于數(shù)據(jù)集本身的原因,下游分析的目的會(huì)有不同的選擇,但毫無疑問最好的填補(bǔ)方法將能夠以較低的計(jì)算要求有效區(qū)分技術(shù)噪聲零值和生物零值(Jiang 等,2022a;Wen 等,2022)。
4、特征選擇
為了降低數(shù)據(jù)維數(shù)以提升計(jì)算分析效率、減少技術(shù)噪聲干擾和模型過擬合的風(fēng)險(xiǎn),我們常常采取特征選擇策略,選取不同細(xì)胞中差異較大的基因,而非整個(gè)數(shù)據(jù)集基因進(jìn)行聚類等后續(xù)分析(Brennecke 等,2013;Jackson and Vogel,2022;Svensson 等,2017)。
在 bulk RNA-seq 分析中,尋找差異基因的方法通常包括基于倍數(shù)變化(FC)的方法、基于統(tǒng)計(jì)檢驗(yàn)的方法和 FC- 統(tǒng)計(jì)檢驗(yàn)方法,顯然后者的篩選結(jié)果和可信度最好(Chung and Storey,2015)。
早期的單細(xì)胞特征選擇方法缺乏平均表達(dá)量與方差之間的校正,導(dǎo)致結(jié)果中高表達(dá)基因的比例過高(Brennecke 等,2013)。EDGE 采用大量弱學(xué)習(xí)器的集成學(xué)習(xí)方法來學(xué)習(xí)細(xì)胞間相似性概率,提取基于信息熵的顯著貢獻(xiàn)作為高可變基因(Sun 等,2020c)。同樣,SAIC 基于迭代聚類最終輸出實(shí)現(xiàn)了最優(yōu)細(xì)胞簇分離(Yang 等,2017)。近期,一些新的特征提取策略被提出并證明了其穩(wěn)定性和有效性,但它們之間的性能權(quán)威驗(yàn)證尚缺乏:包括基于基因表達(dá)分布矩陣的方法 SCMER(Liang 等,2021b)、RgCop(Lall 等,2021)、scPNMF(Song 等,2021a)、SIEVE(Zhang 等,2021e);基于熵的方法 IEntropy(Li 等,2022g)、infohet(Casey 等,2023);綜合考慮聚類的方法有 Triku(Ascensión 等,2022)、FEAST(Su 等,2021)等。由于上述方法絕大多數(shù)忽略了整體的依賴于基因表達(dá)的特征,因此提出了綜合的方法,如 Triku 使用 k 最近鄰圖的方法對(duì)基因表達(dá)模式進(jìn)行綜合探索和分類,實(shí)現(xiàn)無偏差地篩選出更有生物學(xué)意義的特征基因;FEAST 在共識(shí)聚類上通過 f 檢驗(yàn)對(duì)特征進(jìn)行排序,并基于特征評(píng)估算法提取 HVG(Wang 等,2022c)。
其他一些方法使用高可變基因以外的特征來表示數(shù)據(jù)集,例如 scVEGs 和 scSensitiveGeneDefine 方法,使用高變異系數(shù)(CV)作為特征提取;BASiCS 方法利用 spike-in 基因的信息(Chen 等,2016b;Chen 等,2021b)。總體來看,基于準(zhǔn)確性、生物學(xué)可解釋性等角度,當(dāng)前特征選擇的主要目標(biāo)是有效提取 HVG,以便對(duì)高維轉(zhuǎn)錄組數(shù)據(jù)進(jìn)行有效的下游分析。
5、降維
由于單細(xì)胞轉(zhuǎn)錄組通常包含數(shù)萬個(gè)甚至更多的基因,不利于直接提取有效信息,在實(shí)際分析過程中,通常需要對(duì)原始測序數(shù)據(jù)進(jìn)行降維。除了利用前文提到的特征選擇方法處理高維單細(xì)胞轉(zhuǎn)錄組測序數(shù)據(jù)外,降維也是一種有效的方法,根據(jù)降維策略可分為線性降維(基于潛在狄利克雷分配(LDA)的方法、基于 PCA 的方法)和非線性降維(基于 t -SNE 的方法、基于 UMAP 的方法)(Andrews and Hemberg,2018;Becht 等,2019;Laurens and Hinton,2008;Peres-Neto 等,2005)。
在線性降維中,LDA 和 PCA 是兩種廣泛使用的算法,LDA 從分類最大的角度區(qū)分特征,而 PCA 則從方差最大的角度正交提取主成分。盡管有 JPCDA、LDA-PLS 等改進(jìn)算法,但是 LDA 模型在單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)中的降維效果仍然不是最優(yōu)的(Tang 等,2014 ; Zhao 等,2020)。PCA 是另一種線性變換,Seurat 通常根據(jù)標(biāo)準(zhǔn)差 -PC 圖的拐點(diǎn)或者 PC 的比例檢驗(yàn)結(jié)果 P 值(ScoreJackStraw 函數(shù))來確定 PC 數(shù)量的多少。其他基于 PCA 的降維方法的變體包括 pcaReduce(?urauskien ?和 Yau,2016),GLM-PCA(Townes 等,2019),RPCA(Gogolewski 等,2019),tRPCA(Candès 等,2011),scPCA(Boileau 等,2020),PCAone(Li 等,2022l)。GLM-PCA 將傳統(tǒng) PCA 分析擴(kuò)展到非正態(tài)分布,通過引入指數(shù)家族似然策略直接處理原始矩陣,使 PCA 擺脫正態(tài)化限制,然后使用偏差對(duì)基因?qū)崿F(xiàn)進(jìn)行排序和提取(Collins 等,2002)。ScPCA 使用對(duì)比 PCA 和稀疏 PCA 分別去除技術(shù)噪音和數(shù)據(jù),進(jìn)一步增加了 PCA 的穩(wěn)定性(Abid 等,2018 ; Zou 等,2006)。由于大多數(shù) scRNA-seq 數(shù)據(jù)集難以通過簡單的線性降維進(jìn)行有效表示,解決這一問題的第一個(gè)策略是基于快速 PCA 分析方法。PCAone 提出了一種新的快速隨機(jī)奇異值分解(RSVD)策略,在 35 分鐘內(nèi)完成 130 萬小鼠腦細(xì)胞單細(xì)胞數(shù)據(jù)的分析(Li 等,2022l)。
非線性降維是另一種解決方案,如非參數(shù)降維方法 t -SNE 和 UMAP,都需要預(yù)先設(shè)置聚類的超參數(shù);而在分類效果上,前者傾向于離散數(shù)據(jù)中細(xì)胞的形成。在合理使用參數(shù)設(shè)定的情況下,UMAP 與 t -SNE 并無明顯差異,即在使用相同的信息初始化方法后,二者可以在保留數(shù)據(jù)集全局結(jié)構(gòu)的同時(shí),產(chǎn)生近似的分析效率(Do and Canzar,2021;Kobak and Linderman,2021)。針對(duì) t -SNE 的改進(jìn)方法包括 net-SNE、qSNE、FItSNE、聯(lián)合 t -SNE(Cho 等,2018a;Linderman 等,2019;Wang 等,2022b),而 UMAP 的改進(jìn)主要來自于 Leland McInnes 課題組對(duì)該方法的自我改進(jìn)(McInnes 等,2018)。為了更好地可視化 t -SNE 或 UMAP 的降維結(jié)果,Hyunghoon Cho 提出了基于局部半徑依賴優(yōu)化的轉(zhuǎn)錄組變異信息 den-SNE/densMAP 方法,以迭代優(yōu)化傳統(tǒng) t -SNE/UMAP 的功能;Stefan Canzar 提出了 j -SNE/jUMAP 來改善多模態(tài)組學(xué)數(shù)據(jù)聯(lián)合可視化結(jié)果,減少可視化的誤導(dǎo)性(Do and Canzar,2021;Narayan 等,2021)。
6、聚類
在單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析中,通過聚類將細(xì)胞劃分為亞群,從而能夠表征多細(xì)胞生物中不同細(xì)胞類型,這有助于我們從細(xì)胞異質(zhì)性的角度準(zhǔn)確地分析不同的組織或發(fā)育過程。聚類的實(shí)際效果會(huì)受到數(shù)據(jù)預(yù)處理步驟的影響,例如浴效應(yīng)歸一化、歸納、降維等。
在特征基因選擇和降維之后,絕大多數(shù)單細(xì)胞是基于距離進(jìn)行聚類的。K 均值聚類算法的概念被用于 SCUBA、SC3 和 RaceID 等應(yīng)用(Grün 等,2015;Kiselev 等,2017;Macqueen, 1967;Marco 等,2014)。在參數(shù)選擇改進(jìn)方面,SAIC 通過 Davies-Bouldin 指數(shù)迭代優(yōu)化多個(gè)初始中心 K 和 P 值,以獲得最優(yōu)解;LAK 將參數(shù)選擇算法應(yīng)用于數(shù)據(jù)集,實(shí)現(xiàn)參數(shù)的自動(dòng)選擇(Davies and Bouldin, 1979;Hua 等,2020;Yang 等,2017)。在超高維數(shù)據(jù)的操作中,LAK 添加 Lasso 懲罰項(xiàng)進(jìn)行標(biāo)準(zhǔn)化,mbkmeans 使用小批量 k 均值實(shí)現(xiàn)百萬細(xì)胞級(jí)別的快速聚類(Hicks 等,2021)。SMSC 應(yīng)用譜聚類方法來提高聚類性能,但對(duì)于超高維數(shù)據(jù)會(huì)損失一定的準(zhǔn)確性(Qi 等,2021)。另一大類廣泛使用的距離聚類方法依賴于共享最近鄰圖結(jié)構(gòu)和圖聚類,其中使用最廣泛的是 Louvain 或 Leiden(Blondel 等,2008;Xu and Su, 2015)。稀有細(xì)胞的識(shí)別需要結(jié)合特定方法進(jìn)行改進(jìn),例如 dropClust 使用局部敏感哈希工作流篩選最近鄰,然后是 Louvain 聚類,它使用指數(shù)衰減函數(shù)來保留更多稀有細(xì)胞的轉(zhuǎn)錄組特征(Sinha 等,2018)。其他基于距離的聚類方法使用不同的算法核心:SIMLR 使用高斯核學(xué)習(xí)模型為數(shù)據(jù)集中潛在的 C 細(xì)胞群體構(gòu)建核矩陣,而 Conos 提出聯(lián)合相互最近鄰(mNN)圖聚類來實(shí)現(xiàn)對(duì)多個(gè)不同單細(xì)胞轉(zhuǎn)錄組樣本的整合分析(Barkas 等,2019 ; Wang 等,2017a)。基于密度的聚類利用樣本分布的接近程度進(jìn)行聚類,DBSCAN 是最經(jīng)典的算法(Ester 等,1996 ; Fukunaga and Hostetler, 1975)。對(duì)于單細(xì)胞測序,densityCut 和 FlowGrid 就是基于此原理設(shè)計(jì)的(Ding 等,2016 ; Fang and Ho, 2021)。層次聚類是一種自下而上的聚類方法,通過無監(jiān)督學(xué)習(xí),不斷重復(fù)計(jì)算細(xì)胞與細(xì)胞的相似性進(jìn)行分類,直至完成預(yù)設(shè)的聚類數(shù)(Guo 等,2015)。隨后,RCA 聚類采用常規(guī)的層次聚類方法,對(duì)映射到全局參考面板的細(xì)胞進(jìn)行聚類;HGC 在 SNN 圖上構(gòu)建層次樹(Li 等,2017;Zou 等,2021)。為了解決常規(guī)層次聚類方法難以對(duì)某一組細(xì)胞進(jìn)行聚類、只允許同一組特征基因進(jìn)行聚類的缺陷,K2Taxonomer 采用約束 K 均值算法擴(kuò)展到樣本組,基于多個(gè)基因集遞歸進(jìn)行積分計(jì)算,以捕獲各種分辨率下的亞組(“類似分類學(xué)的細(xì)胞”)(Reed and Monti, 2021)。Mrtree 將層次聚類的策略應(yīng)用于平面簇的多個(gè)劃分,并構(gòu)造多分辨率協(xié)調(diào)樹用于細(xì)胞聚類(Peng 等,2021a)。最近,Zelig 和 Kaplan(2020)提出了一種 KMD 聚類方法,通過平均鏈接層次聚類模型在聚類時(shí)消除了超參數(shù) K,大大減少了主觀性帶來的判斷誤差。
深度學(xué)習(xí)聚類方法是將機(jī)器學(xué)習(xí)方法與上述單細(xì)胞轉(zhuǎn)錄組聚類策略相結(jié)合,可以以無監(jiān)督、監(jiān)督或半監(jiān)督的形式實(shí)現(xiàn)更高效的聚類結(jié)果。這些方法傾向于學(xué)習(xí)一種非線性變換,通過將原始高維數(shù)據(jù)映射到較小的潛在空間中來獲得最佳的低維表示。總體而言,這種方法避免了傳統(tǒng)聚類方法對(duì)聚類前數(shù)據(jù)處理方法選擇的影響。無監(jiān)督聚類方法包括 ADClust、DESC、SAUCIE、VAE-SNE 等,通常不需要預(yù)設(shè)聚類個(gè)數(shù)等參數(shù),以自主學(xué)習(xí)的方式完成對(duì)數(shù)據(jù)集的分析處理(Amodio 等,2019;Graving and Couzin,2020;Li 等,2020c;Zeng 等,2022c)。雖然無監(jiān)督聚類方法避免了手動(dòng)輸入聚類個(gè)數(shù)等參數(shù),可以延伸到超高維細(xì)胞聚類,但有時(shí)利用高質(zhì)量標(biāo)注數(shù)據(jù)集或其他先驗(yàn)知識(shí)輔助約束進(jìn)行監(jiān)督或半監(jiān)督聚類,可以實(shí)現(xiàn)更為準(zhǔn)確的細(xì)胞類型分類,提高聚類性能(Bai 等,2021)。基于遷移學(xué)習(xí)的 ItClust、基于互監(jiān)督 ZINB 自編碼器和圖神經(jīng)網(wǎng)絡(luò)(GNN)的 scDSC、基于軟 K 均值卷積自編碼器的 ScCAE、基于 Cramer-World 距離最大均值懲罰高斯混合自編碼器的 SeGMA、基于時(shí)間序列聚類網(wǎng)絡(luò) STCN 都是廣泛使用的監(jiān)督聚類(Gan 等,2022 ; Hu 等,2022a ; Hu 等,2020a ; Ma 等,2021b ; Smieja 等,2021)。此外,Zhang 團(tuán)隊(duì)(Yang 等,2023b)利用分層 GAN 設(shè)計(jì)了另一種廣泛使用的深度學(xué)習(xí)方法 IMDGC,用于單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析,以生成的方式構(gòu)建細(xì)胞嵌入簇。
針對(duì)聚類中的特殊情況,設(shè)計(jì)了有針對(duì)性的聚類方法:GiniClust(Jiang 等,2016)(更新為 GiniClust 3(Dong and Yuan,2020)、MicroCellClust(Gerniers 等,2021)用于稀有細(xì)胞亞群聚類;EDClust(Wei 等,2022)、ENCORE(Song 等,2021b)和 MLG(Lu 等,2021)用于降噪和消除批次效應(yīng);ClonoCluster(克隆起源信息)(Richman 等,2023)、IsoCell(可變剪接信息)(Liu 等,2023)使用附加信息進(jìn)行聚類。Wu 和 Yang 從特征選擇對(duì)聚類的影響的角度對(duì)聚類方法進(jìn)行了評(píng)估,他們證明更具代表性的特征選擇會(huì)提高細(xì)胞聚類的水平,基于“聚類相似性”的方法(我們綜述中提到的大多數(shù)基于距離的聚類方法)通常具有廣泛的高聚類類型性能;然而,高精度和高運(yùn)行速度需要根據(jù)實(shí)際數(shù)據(jù)集進(jìn)行有針對(duì)性的選擇(Su 等,2021;Yu 等,2022)。雙重浸入 (double dipping) 是一個(gè)顯著的問題,即在細(xì)胞聚類和差異表達(dá)基因中使用相同的表達(dá)數(shù)據(jù),導(dǎo)致在細(xì)胞聚類不正確時(shí) DE 基因的錯(cuò)誤發(fā)現(xiàn)率 (FDR) 過高。例如,如果只存在一個(gè)特定的細(xì)胞聚類,則不應(yīng)將任何基因視為差異基因。為了系統(tǒng)地解決這個(gè)問題,ClusterDE 采用了聚類對(duì)比學(xué)習(xí)策略進(jìn)行聚類后 DE 測試。該方法與截?cái)嗾龖B(tài)分布 (TN) 檢驗(yàn)和 Countsplit 方法相比,在不同閾值范圍內(nèi)具有更好的 FDR 控制 (Song 等,2023a)。
7、細(xì)胞類型注釋
細(xì)胞分型注釋是指利用特定的信息對(duì)單細(xì)胞測序數(shù)據(jù)集中的細(xì)胞或細(xì)胞亞群進(jìn)行注釋,作為后續(xù)生物學(xué)分析的基礎(chǔ)。最常用的策略是對(duì)細(xì)胞進(jìn)行無監(jiān)督聚類,然后根據(jù)標(biāo)記基因進(jìn)行注釋,例如 scCATCH、SCSA (Cao 等,2020b;Shao 等,2020a),但它難以處理復(fù)雜的高維數(shù)據(jù)集 (Franzén 等,2019;Luecken and Theis, 2019;Zhang 等,2019c)。目前已經(jīng)開發(fā)了多種自動(dòng)細(xì)胞分型方法,大致可分為兩類,即依賴參考和無參考的注釋方法。
依賴參考信息的注釋方法要求用戶提供預(yù)先注釋的高質(zhì)量單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)集或來自 PanglaoDB 數(shù)據(jù)庫、ScType 數(shù)據(jù)庫等的先驗(yàn)知識(shí)進(jìn)行比對(duì)(Ianevski 等,2022)。根據(jù)方法原理的不同,可分為基于層次樹的方法(CHETAH(de Kanter 等,2019)、Garnett(Pliner 等,2019)、HieRFIT(Kaymaz 等,2021)、scHPL(Michielsen 等,2021)、scMRMA(Li 等,2022e)、基于相似性的方法(SingleR(Aran 等,2019)、scmap(Kiselev 等,2018)、deCS(Pei 等,2023)、scID(Boufea 等,2020)、scMatch(Hou 等,2019)、Symphony(Kang 等,2021)、基于簽名基因的方法(Cellassign(Zhang 等,2021)、基于特征基因的方法(Cellassign(Zhang 等,2022)。al., 2019a )、Cell-ID(Cortal 等,2021)、scMAGIC(Zhang 等,2022g)、SciBet(Li 等,2020b)和其他 DL 方法。作為早期方法,ACTINN 是一種使用 3 個(gè)隱藏層神經(jīng)網(wǎng)絡(luò)進(jìn)行注釋分類的深度學(xué)習(xí)方法(Ma and Pellegrini, 2020)。SCPred 隨后提出了一種基于嵌入的無偏特征選擇的機(jī)器學(xué)習(xí)概率預(yù)測方法(Alquicira-Hernandez 等,2019)。其他方法如 Seurat 在 PCA 空間中投影查詢細(xì)胞并通過加權(quán)投票分類器訓(xùn)練細(xì)胞分型注釋;scSorter 采用高斯混合模型,GraphCS 使用虛擬對(duì)抗訓(xùn)練 (VAT) 損失修改的 GNN 來擴(kuò)展到多物種、大規(guī)模細(xì)胞注釋數(shù)據(jù)集(Guo and Li,2021;Zeng 等,2022a)。
不依賴參考信息的注釋方法使用預(yù)先訓(xùn)練的深度學(xué)習(xí)模型,可以直接使用查詢數(shù)據(jù)集作為輸入進(jìn)行細(xì)胞分類。scDeepSort 使用來自人類細(xì)胞圖譜 (HCL) 和小鼠細(xì)胞圖譜 (MCA) 數(shù)據(jù)庫的單細(xì)胞圖譜作為預(yù)訓(xùn)練加權(quán) GNN 模型的輸入,該模型適用于人類和小鼠細(xì)胞注釋并取得良好的效果(Han 等,2018b;Han 等,2020;Shao 等,2021b)。類似地,Pollock 是一個(gè)預(yù)訓(xùn)練的人類癌癥參考 VAE 模型,用于對(duì)癌癥環(huán)境中的多模態(tài)細(xì)胞進(jìn)行分類(Storrs 等,2022)。雖然使用起來更方便,但對(duì)于差異顯著的查詢數(shù)據(jù)集難以達(dá)到更好的細(xì)胞注釋效果,而且由于準(zhǔn)確性和預(yù)訓(xùn)練參考數(shù)據(jù)集的數(shù)量也難以擴(kuò)展應(yīng)用。還有一些其他用于有針對(duì)性領(lǐng)域研究的細(xì)胞注釋工具,例如,用于人類腎細(xì)胞注釋的 DevKidCC(Wilson 等,2022),用于識(shí)別癌癥和正常細(xì)胞的 ikarus(Dohmen 等,2021)。總體而言,無參考注釋方法的性能受到預(yù)訓(xùn)練參考數(shù)據(jù)集的覆蓋率和準(zhǔn)確性的制約。
目前,改進(jìn)細(xì)胞注釋工具以在大平臺(tái)和多細(xì)胞模式下統(tǒng)一分配細(xì)胞類型是細(xì)胞注釋研究的主流方向,最新的 Cellar 和 ELeFHAnt 方法在這方面做了一些嘗試并取得了初步成果(Hasanaj 等,2022 ; Thorner 等,2021)。總體而言,基于相似性的注釋方法計(jì)算量大,在應(yīng)用于非常大的查詢和參考數(shù)據(jù)集時(shí),往往會(huì)在準(zhǔn)確率和速度之間做出權(quán)衡,因此一般只適合在較小的數(shù)據(jù)集中進(jìn)行細(xì)胞分類;對(duì)于較大規(guī)模的數(shù)據(jù)集,建議使用 F 檢驗(yàn)特征選擇或 MLP 分類器(Hu 等,2020a ; Huang and Zhang, 2021 ; Ma 等,2021c)。此外,半監(jiān)督遷移學(xué)習(xí)的方法,如 Itclust,在發(fā)現(xiàn)新的細(xì)胞亞型方面也有不錯(cuò)的效果。近年來,基于上述參考注釋方法分類的新方法不斷完善,VAE 等深度學(xué)習(xí)模型也在該領(lǐng)域得到應(yīng)用。
8、差異表達(dá)分析(DEG)
統(tǒng)計(jì)檢驗(yàn)是 Bulk RNA-seq 的差異基因分析中常用到的方法,類似章節(jié) 2.4HVG Selection 算法:通常以 P 值和對(duì)數(shù)倍變化量作為重要參數(shù)。統(tǒng)計(jì)檢驗(yàn)包括 t 檢驗(yàn)(兩個(gè)樣本為基礎(chǔ)),Wilcoxon 檢驗(yàn),Kolmogorov-Smirnov 檢驗(yàn)(KS 檢驗(yàn)),Kruskal-Wallis 檢驗(yàn)(KW 檢驗(yàn)),其中一些在單細(xì)胞轉(zhuǎn)錄組 DEGs 的檢驗(yàn)中也被廣泛使用。基于此,發(fā)展了相應(yīng)的檢測工具:limma(Ritchie 等,2015),edgeR(Robinson 等,2010),DESeq2(Love 等,2014)。limma 和 edgeR 算法均由 Smyth GK 提出,前者基于正態(tài)或近似正態(tài)分布模型,后者基于過度離散的泊松分布模型。DESeq2 基于 NB 分布模型進(jìn)行假設(shè)檢驗(yàn),對(duì) DEG 采用經(jīng)驗(yàn)貝葉斯程序。目前 limma 由于特定的分布模型假設(shè),在 RNA 計(jì)數(shù)分析中誤差較大,雖然 edgeR 和 DESeq2 都利用貝葉斯模型對(duì)過度離散進(jìn)行歸一化,但后者通過數(shù)據(jù)集 reads 的平均值和異常值檢測促進(jìn)了 CPM 閾值的篩選,分析效果更好。
單細(xì)胞轉(zhuǎn)錄組 DEG 按照時(shí)間和分析方法大致可以分為早期零值參數(shù)檢驗(yàn)、非參數(shù)檢驗(yàn)和其他方法。由于 scRNA-seq 數(shù)據(jù)中存在大量零數(shù),早期的方法大多基于此觀察做參數(shù)檢驗(yàn),例如 Monocle (Trapnell 等,2014)、SCDE (Kharchenko 等,2014)、MAST (Finak 等,2015)、scDD (Korthauer 等,2016)、D3E (Delmans and Hemberg, 2016)、TASC (Jia 等,2017)、DEsingle (Miao 等,2018) 和 HIPPO (Kim 等,2020b)。對(duì)以上一些方法的評(píng)測表明,雖然它們在單細(xì)胞數(shù)據(jù)集的分析中普遍取得了不錯(cuò)的效果,但對(duì)于批量數(shù)據(jù)(Soneson and Robinson, 2018)相比 DEA 方法并沒有明顯的性能提升。對(duì)于不同的數(shù)據(jù)集,有可能沒有最好的分布模型,因此一種替代解決方案是考慮非參數(shù) DEA 方法。
非參數(shù)檢驗(yàn)或無分布檢驗(yàn)不需要對(duì)數(shù)據(jù)分布形式做事先假設(shè),因此適用于多數(shù)據(jù)集的分析,常用方法有 Swish(Zhu 等,2019a)、IDEAS(Zhang 等,2022d)、ccdf(Gauthier 等,2021)、distinct(Tiberi 等,2022)。Swish 通過 Salmon Gibbs 評(píng)估轉(zhuǎn)錄本水平,然后用 Mann-Whitney Wilcoxon 檢驗(yàn)計(jì)算 FC 值。IDEAS 是一種使用 Jensen-Shannon 散度(JSD)或 Wasserstein 距離(Was)進(jìn)行基因差異表達(dá)測量的偽 F 統(tǒng)計(jì)量檢驗(yàn),P 值由基于 PERMANOVA 的距離測試器基于核的回歸生成。Ccdf 是一種依賴條件累積分布函數(shù)的條件獨(dú)立性檢驗(yàn),通過多元回歸模型預(yù)測 DEG。Distinct 提出了一種分層非參數(shù)置換方法,使用經(jīng)驗(yàn)累積分布函數(shù) (ECDF) 的總距離進(jìn)行 DEG 識(shí)別。替代方法包括深度學(xué)習(xí)策略 MRFscRNAseq (Li 等,2021a)、基于擬時(shí)序推斷的 PseudotimeDE (Song and Li, 2021)、基于非預(yù)聚類的 singleCellHaystack (Vandenbon and Diez, 2020)、基于多重評(píng)分的 MarcoPolo (Kim 等,2022)。建議不同的單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)集應(yīng)采用數(shù)據(jù)特定的 DEGs 檢測策略,以優(yōu)化 DEGs 分析,基于 scCODE 工作流程,可以使用涉及 CDO(DE 基因順序)和 AUCC(一致性曲線下面積)的指標(biāo)找到最優(yōu)化的 DEGs 方法(Zou 等,2022)。此外,研究方法在不同的研究背景下會(huì)有特定的研究取向,例如在給藥后的劑量反應(yīng)研究中,DEGs 分析、LRT 線性檢驗(yàn)和貝葉斯多組檢驗(yàn)均比其他方法有更好的結(jié)果(Nault 等,2022)。
9、可視化
單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析可視化是指將上述分析結(jié)果以圖形的形式直觀地呈現(xiàn),ggplot2 是 R 中最廣泛的可視化工具,在 R 中被廣泛使用,可以大大增強(qiáng)繪圖能力(Wickham,2009)。ARL 是另一個(gè)專門顯示標(biāo)記基因關(guān)聯(lián)圖并可顯示其在每個(gè)簇中的特征的 R 包(Gralinska 等,2022)。此外,還有其他專門用于標(biāo)記基因可視化的包,如 Complex Heatmap,本文不再詳細(xì)介紹。HVG 可視化通常以火山圖的形式呈現(xiàn),默認(rèn)情況下,圖的左側(cè)和右側(cè)部分分別是代表性不足的基因和代表性過高的基因,而中間是恒定基因。Enhanced Volcano 是一個(gè)專門用于繪制火山圖的 R 包,默認(rèn)情況下也可以使用 ggplot2 來獲得更好的結(jié)果。簇可視化通常以 PCA 圖、t-SNE 圖和 UMAP 圖呈現(xiàn),但值得注意的是,可視化的結(jié)果非常具有欺騙性,因?yàn)橐恍┬〉募?xì)胞亞群可能代表 UMAP 圖中顯示的大量細(xì)胞。為了解決這些問題,提出了 den-SNE/densMAP、j-SNE/j-UMAP 等改進(jìn)方法(Macqueen,1967;Marco 等,2014)。此外,F(xiàn)astProject 可以輸出注釋簇的 2D 顯示,PieParty 可以在簇 2D 圖中為每個(gè)基因繪制顏色圖(DeTomaso and Yosef,2016;Kurtenbach 等,2021)。
同時(shí),單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)的交互式可視化是目前的熱門領(lǐng)域,諸如 Single Cell Explorer 等軟件可以一定程度上實(shí)現(xiàn)交互式可視化,但仍需增加交互自由度,以提供更全面的單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù) 3D 呈現(xiàn)(Cakir 等,2020;Feng 等,2019)。為此,CellexalVR 利用 VR 理論進(jìn)行交互可視化;CellView 是一個(gè)基于 Web 的工具,包括用于不同用途的探索選項(xiàng)卡、共表達(dá)選項(xiàng)卡、子簇分析選項(xiàng)卡模塊;Cellxgene VIP 是一個(gè)基于 cellxgene 框架的插件,并擴(kuò)展到基于多個(gè)模塊組合的 ST 數(shù)據(jù)的交互式可視化(Bolisetty 等,2017 ; Legetth 等,2021 ; Li 等,2022f)。
10、單細(xì)胞模擬
隨著單細(xì)胞轉(zhuǎn)錄組方法的不斷擴(kuò)展,基準(zhǔn)測試成為了重要挑戰(zhàn),關(guān)鍵問題是需要穩(wěn)定可靠的數(shù)據(jù),因?yàn)閱渭?xì)胞轉(zhuǎn)錄組的直接測序可能缺乏基本事實(shí)。真實(shí)的單細(xì)胞模擬數(shù)據(jù)為基準(zhǔn)測試提供了已知的事實(shí),允許使用真實(shí)數(shù)據(jù)進(jìn)行訓(xùn)練,同時(shí)匹配實(shí)際數(shù)據(jù)的特征。此外,模擬數(shù)據(jù)比真實(shí)數(shù)據(jù)提供了更大的靈活性,使分析師能夠根據(jù)特定的測試方法調(diào)整諸如 dropout rate 等參數(shù)。
Splatter 是一個(gè)兩步模擬框架,首先模擬來自真實(shí)數(shù)據(jù)的估計(jì)參數(shù),然后合并來自用戶的額外參數(shù)(Zappia 等,2017)。其六個(gè)預(yù)先設(shè)計(jì)的管道模塊接口確保了數(shù)據(jù)生成的可重復(fù)性。最近的更新側(cè)重于專業(yè)化和泛化。在專業(yè)化領(lǐng)域,splaPop 生成具有遺傳效應(yīng)(數(shù)量性狀基因座)的人口規(guī)模數(shù)據(jù),而 dyngen 模擬動(dòng)態(tài)細(xì)胞過程,如發(fā)育軌跡(Azodi 等,2021;Cannoodt 等,2021)。在泛化領(lǐng)域,Li 的團(tuán)隊(duì)介紹了理想模擬的六個(gè)概念,包括真實(shí)性、基因的保存、基因相關(guān)性的捕獲、穩(wěn)健性、參數(shù)可調(diào)性和效率(Song 等,2023b;Sun 等,2021)。隨后,scDesign2 提出來滿足所有 6 個(gè)屬性(Sun 等,2021),接著是 scDesign3,解決單細(xì)胞組學(xué)統(tǒng)計(jì)模擬的空白(Song 等,2023b)。模擬準(zhǔn)確性和透明度的提高增強(qiáng)了不同單細(xì)胞數(shù)據(jù)處理方法之間的基準(zhǔn)測試,指導(dǎo)選擇最合適的方法以滿足特定數(shù)據(jù)和許可需求。
